
公司动态
2024-2025年,全球主要医疗器械市场的监管规则正在经历深刻变革。对中国出海企业而言,这些变化既是必须跨越的合规门槛,也是重新定义竞争格局的关键变量。

核心变化
1
质量体系法规(QMSR)与ISO 13485整合
2026年2月2日生效,要求建立覆盖全生命周期的风险管理体系。
2
网络安全要求从指南升级为强制法规
2025年6月生效,含软件/固件的器械必须在新品设计阶段融入安全控制。
3
UDI分阶段强制实施
Ⅱ类器械需在2024年9月24日前完成GUDID数据库注册,I类器械截止期为2025年9月24日。
4
境外飞行检查力度加大
对海外工厂实施突击检查,拒绝检查将面临进口禁令。
冲击与应对
1
合规成本显著增加
FDA用户费用在2025财年上调,企业需提前进行财务规划(510(k)标准费用从21,760美元涨至24,335美元,涨幅11.8%,年度注册费从7,653美元涨至9,280美元,涨幅超过20%。
2
系统性能力建设
建议立即启动质量体系与ISO 13485的差距分析,引入专业网络安全顾问进行威胁建模。
3
供应链韧性考量
探索在墨西哥等地设厂以满足“本地化”要求,分散单一市场风险。
关键时间节点
1
Ⅲ类及部分IIb类器械
过渡期最长可延至2027年。
2
IIa类及I类器械
过渡期最长可延至2028年。
前提条件:器械设计未发生重大变化,且制造商已在2024年3月31日前与公告机构签订MDR认证合同。
技术文件要求升级
1
临床证据要求更严格
Ⅲ类和高风险IIb类器械需根据上市后临床跟踪数据持续更新临床评估报告。
2
文件完整性成瓶颈
75%制造商首次提交的技术文件完整性低于50%。
3
UDI全面实施
隐形眼镜Master UDI-DI规则于2025年11月9日起正式实施。
市场影响
1
公告机构资源紧张
虽62%申请能在2个月内完成签约,但总体审核周期仍较长。
2
本地化需求凸显
考虑在欧盟设立本地公司或与有实力的授权代表建立深度合作。
认证路径细分
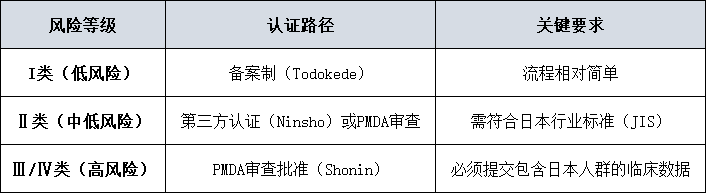
核心挑战
1
临床数据缺口
PMDA对包含亚洲人种(尤其是日本人群)的临床数据愈发重视。
2
质量管理体系本地化
需符合日本版QMS省令,包含额外的供应商管理和上市后监督要求。
3
语言与沟通壁垒
技术文件需为日文版本,与PMDA的高效沟通至关重要。
法规更新亮点
1
基本安全与性能要求(ESPR)更新
RDC 848/2024取代旧法规,强调全生命周期风险管理。
2
简化注册流程
IN 290/2024为已获澳、加、美、日批准的Ⅲ/Ⅳ类器械建立优化程序。
3
强制性本地代表(BRH)
所有外国制造商必须指定巴西注册持有人。
认证周期现实
1
I类器械
备案通知约需30天。
2
Ⅱ类器械
Cadastro路径需3-6个月。
3
Ⅲ/Ⅳ类器械
注册路径可能长达8-12个月,需进行巴西GMP审核。
额外认证要求
1
INMETRO认证
有源医疗器械及部分无源器械强制性要求。
2
ANATEL认证
所有带通讯功能的设备必须办理。
法规变化
1
唯一设备标识(UDI)强制实施
2025年3月24日法规生效,按风险等级分阶段执行。
2
软件类医疗器械规范明确
含AI医疗软件的监管分类、算法验证要求细化。
3
特定产品重新分类
处方眼镜镜片多数豁免注册,但治疗性镜片仍需注册。
应对策略
1
尽早启动UDI合规
按产品风险分类制定合规时间表,避免截止期前扎堆申报。
2
重新评估软件产品
依据TGA最新指南评估分类和合规路径,充分利用过渡期。
3
强化全生命周期管理
将上市后监督、不良事件监测深度融入质量管理体系。
认证路径优化
1
“认证杠杆”效应
获得FDA或MDR认证后,可利用巴西、澳大利亚等地的简化程序加速市场准入。
2
资源优先级排序
根据产品特性和目标市场规模,制定差异化的认证战略。
合规成本管控
1
前期投入评估
认证费用、临床评价成本和时间成本需纳入产品出海总预算。
2
长期维护考量
上市后监管、定期报告等持续合规成本不容忽视。
本地化深度
1
超越法律要求
在欧盟、日本等市场,临床数据本地化已成为竞争优势而非单纯合规要求。
2
合作伙伴选择
经验丰富的本地代表、授权代表可显著提升合规效率。
全球主要市场的监管变革正在重塑医疗器械出海的游戏规则。对企业而言,深入理解这些变化并制定前瞻性的合规战略,已成为在激烈国际竞争中脱颖而出的关键所在。
盛恩医药-注册事务
盛恩医药,精通各国认证法规,熟悉各国认证流程,注册事务具体服务包括:欧盟CE、美国FDA、巴西ANVISA、国内NMPA(分类界定、属性界定、创新申报、型检、临床方案CRO、同品种CER、产品注册)以及其他北美、欧洲、南美、东南亚、亚太地区法规注册认证咨询及定制化服务。
针对国产医疗器械的“走出去”需求,盛恩医药根据不同的海外市场,针对不同企业的不同开发阶段、不同产品的情况,可提供定制式解决方案,帮助企业顺利出海。
针对进口产品寻求中国上市支持需求,基于中国的政策法规及盛恩医药的经验积累,可为进口产品提供全流程服务,帮助进口产品顺利上市。


关于我们
盛恩医药是一家专注于医疗器械注册咨询及临床试验服务的CRO组织,提供CRO、SMO、第三方稽查、独立影像评估服务、FA咨询等服务。公司核心团队成员均具有十年以上临床试验项目服务经验及注册咨询服务经验,完成数百项临床试验项目,并成功获得注册证,特别是心血管领域、外周血管领域、骨科领域、神经介入领域、消化科领域、普外科领域、口腔领域、整形美容领域有着丰富的项目经验。公司秉承“诚信服务、合作共赢”的理念,努力提升服务意识,加强医学能力培养,构建完善的项目管理体系,致力成为行业领先的CRO。
相关业务咨询电话
李老师 18790512921


中华医学会 | 中国医师协会 | 中国临床试验注册中心 | 中华人民共和国科技部 | ClinicalTrials | NMPA | 临床试验机构备案管理信息系统